來源:MedTrend 醫趨勢??? ? ? ? 發表日期:2018年6月5日
? ? ? ?FDA局長Scott Gottlieb從2017年5月走馬上任后,一直在加速藥物審批程序,比如頒布「突破性療法」認證程序將審查時間減少到3個月,縮短將近1/3的時間,2017年新藥審批數創21年來新高,有46種新藥獲批,開啟“以患者為中心”的藥物經濟價值預測時代。
? ? ? ?近日,在芝加哥舉行的美國臨床腫瘤(ASCO)年會上,Scott Gottlieb透露FDA或將頒布重大創新性腫瘤新藥審批新政策——“實時審評制”。腫瘤新藥上市審批或將提前到臨床階段,完成臨床試驗后可直接上市,無需排隊審批。
? ? ? 顛覆式打破抗癌新藥審評監管中的障礙,對于藥企來說無疑是重大利好消息!
▲FDA局長Dr. Scott Gottlieb,圖片來源Cameron Costa | CNBC
什么是“實時審評制”?
? ? ?“實時審評制”的具體流程是根據藥企提供的腫瘤新藥做一份共享申請表格,允許FDA審查員將評論添加到這些背景文件中,確保信息及時溝通和分享。
? ? ? 在提交申請表和被批準之前,FDA需要預先和實時審議臨床試驗數據:
? ? ? ??一是檢查藥企提供的數據完整性和是否齊備;
? ? ????二是打破以往中規中矩的審評流程,幫助藥企解決試驗藥品的質量問題。
? ? ? 也就是說,原先的先進行臨床試驗再進行新藥申請審批,轉為邊進行臨床試驗邊進行新藥審批。
腫瘤新藥審批提速至少半年!惠及所有癌癥新藥
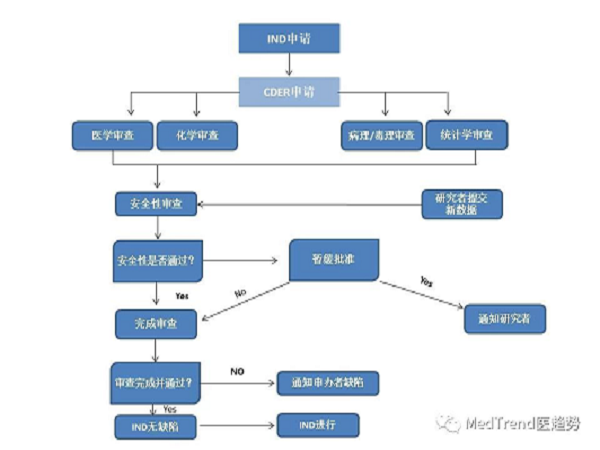
以往新藥研發到上市流程
按照原來新藥研發到上市的流程,可分為:
臨床前研究:研究開發(2-3年)、臨床前試驗(2-4年)
臨床試驗審批(IND):(1個月以上)
臨床試驗(一般3-7年):
人體試驗共分三期:
Ⅰ期臨床 20-100例,正常人,主要進行安全性評價。
Ⅱ期臨床 100-300例,病人,主要進行有效性評價 。
Ⅲ期臨床 300-5000例,病人,擴大樣本量,進一步評價。
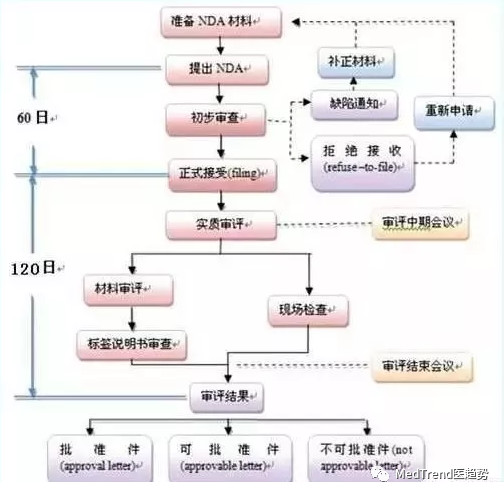
藥上市審批(NDA):半年左右
上市后研究:臨床監測期——Ⅳ期臨床,受試者大于2000例;
上市后再審批(上市后4-10年):重新審核 NDA 中的有效性和安全性。
? ? ? 傳統的藥物審評,80%的審查都集中在臨床部分,20%會專注產品自身的問題。所以,一款腫瘤新藥從研發到上市,期間起碼需要花費十幾年的時間。許多新藥研發在臨床1期、臨床2期就已經失敗,能夠進入臨床3期階段的寥寥無幾。
? ? ? 而原來藥企在做完臨床3期試驗后,才能提交報告去排隊等候FDA審批批復,根據2017年的標準審評時間為17個月左右,而回答問話、補充相應材料都花費大量時間。
“實時審評制”縮減新藥上市時間
? ? ? ? 如果實行“實時審評制”后,藥企在進行臨床試驗階段,FDA就開始參與試驗數據審評,隨時在企業做實驗的過程中告知需要改進之處,或者補充材料及補做試驗,如果數據不合格,或在臨床1期、2期階段就將藥品淘汰,避免藥企后續繁復的試驗和申請工作。
? ? ? ? 而完全臨床3期階段時,藥企或將跳過審批直接上市,對藥企來說,具有顛覆性意義。大大縮短了新藥上市時間,預計至少提速半年時間,也讓腫瘤患者能更快獲得更優質的藥物治療。
目前,“實時審評制”已在上市腫瘤藥擴大適應癥申請中試點應用。如果試點成功,FDA或將會把此審評方法和流程擴展到所有癌癥新藥上市申請程序中去。
? ? ? ?此外,除了腫瘤藥,FDA也有意簡化基因治療和細胞治療的審查流程。
? ? ? ?Scott Gottlieb表示,FDA正在進行細胞和基因治療的新型臨床試驗設計的應用科學研究,并且通過監管制定藥物加快開發計劃。包括使用“突破性療法”認定,以及再生醫學先進療法認定(RMAT)。
FDA多渠道加速審批提速,兩款CAR-T療法從中受益
? ? ? 早在2017年底,FDA就宣布將對特定癌癥藥物審批提速。
? ? ? ??一是對已經獲批的一個適應癥癌癥療法,通過遞交基于“更有針對性的數據集(如單臂研究)”的補充申請,獲得第二個適應癥的批準。
? ? ????二是根據中間臨床終點(intermediate clinical endpoints)加速批準新藥上市。
? ? ? 兩個突破性CAR-T療法:諾華的Kymriah和Kite的Yescarta都分別獲得了FDA優先審評資格和快速通道資格,都成為FDA新藥審批提速的受益者。
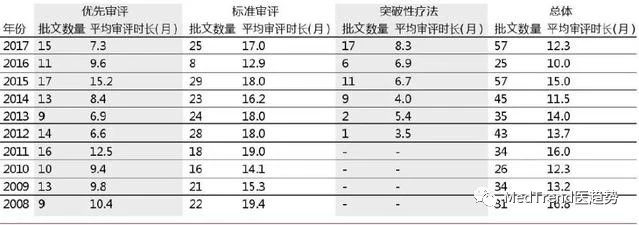
▲不同審評方式時長對比
? ? ?? ?根據上表顯示,2017年標準審評時間為17個月,而進入優先審評的藥物為7.3個月,突破性療法的審評時間為8.3個月。相比標準審評時間減少10個月左右。Kymriah和Yescarta這些優先審評路徑下的藥物獲批時限約為7.3個月,達到2013年以后的最快速度。
? ? ? ? 如果實施“實時審評制”,CAR-T療法連這7.3個月的審評時間或將免去!未來CAR-T療法競爭也將越來越激烈。
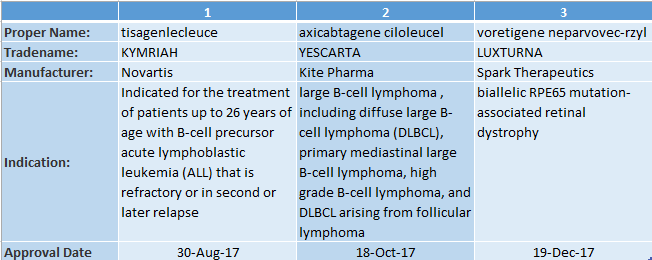
▲FDA批準的3種基因療法(信息來源:FDA官網)
? ? ? ? 在基因療法方面,去年就有三種基因療法獲得批準:?兩種治療血癌基于細胞的基因療法,一種治療遺傳性視網膜萎縮癥的直接型基因療法,將基因療法從理想變為現實。但這三類基因療法產品只是冰山一角。
▲2017年11月發布的再生醫學框架中提到將采取5種途徑加快再生醫學產品審批(圖片來源:FDA官網)
? ? ? ?正如前文所說的,FDA對特定藥物自2016年12月開始,啟動再生先進療法認定(RMAT)。截至2018年4月底,共有62份RMAT認定材料提交,FDA頒發了19個認定。在這19種產品中,有14種也獲得了罕見藥認定。表明該計劃促進了罕見疾藥物的開發。
? ? ? ?麻省理工學院發表的一篇論文基于2017年的930種在研產品線預測。到2022年底,將有約40種基因療法產品獲得FDA批準。此外,這些批準的基因療法中45%都將用于治療癌癥。
一直以來,FDA一面頂著快速批準藥物的壓力,一面希望獲取令人信服的臨床數據,努力在二者之間達成平衡。
? ? ? ?如果FDA“實時審評制”實施,或許有助于實現這種平衡。讓藥物研發在前中期就能基于合理預測的審評進行實時數據調整,幫助新產品批準后能對其安全問題進行更多實時監控。
? ? ? ?未來,腫瘤新藥研發上市速度或將創新高,更多腫瘤新藥和療法的競爭也將越來越激烈。更多新藥的上市,對于也給了患者一個擺脫病魔的更多機會。