? ? ?? 來源:聯川基因
Abstract
? ?? “腫瘤免疫檢查點阻斷”是指使用治療性抗體,破壞負免疫調節檢查點,并激活預先存在的抗腫瘤免疫反應。通過靶向幾種檢查點分子,如細胞毒性T淋巴細胞抗原4 (CTLA4)、程序性細胞死亡蛋白-1 (PD1)和PD1配體1 (PD-L1)的抗體已經在臨床上取得了初步成功,鑒于此,美國食品和藥物管理局(FDA)批準了幾種癌癥類型的多藥制劑。然而,臨床醫生仍然只有非常有限的工具來區分那些對治療有反應和沒有反應的患者。而且越來越多的臨床證據表明,相當比例的最初反應者最終在數月或數年后復發,出現致命的耐藥性疾病。這激起了一波研究腫瘤對免疫檢查點阻斷的內在抵抗的分子機制的浪潮,導致抗腫瘤免疫的關鍵生物學過程的重新發現,即干擾素信號和抗原遞呈。其他的研究努力闡明常規的影響癌癥的免疫信號通路,如WNT-β-catenin信號、細胞周期調控信號,絲裂原激活蛋白激酶信號和腫瘤抑制基因PTEN失活的途徑。這篇2020年1月發表于《Nat Rev Immunol》的文章回顧了這些耐藥的分子機制,以及免疫耐受微環境形成的生物學過程,并探索正在進行的方法來克服對免疫檢查點抑制劑的耐藥性,并擴大可受益于免疫檢查點抑制劑的患者的范圍。
Introduction
? ? ?? 癌癥免疫治療是利用人類免疫系統的細胞毒性潛能,特別是腫瘤特異性細胞毒性T細胞來治療惡性腫瘤的一種策略。在不同類型的癌癥免疫治療中,免疫檢查點阻斷的影響最為廣泛,有幾種針對細胞毒性T淋巴細胞抗原4 (CTLA4)或程序性細胞死亡1 (PD1)和配體1 (PD-L1)的抗體被批準用于多種不同的癌癥。大量針對其他假定的免疫檢查點(如LAG3、TIGIT、TIM3、B7H3、CD39、CD73和腺苷A2A受體)的抗體和小分子干擾腫瘤細胞與T細胞、髓細胞與T細胞之間的負調控,正處于臨床和臨床前的研究中。
? ? ?? 患者內在因素(如年齡、性別、HLA基因型和基因多態性),腫瘤內在因素(如宿主免疫系統和腫瘤基質)和環境因素(如腸道微生物群)都可能導致免疫檢查點阻斷的成功或失敗。然而,腫瘤細胞內在因素(這里定義為腫瘤內在因素),與腫瘤細胞本身的遺傳、轉錄或功能特征有關,是對藥物敏感和耐藥的主要決定因素。腫瘤內在因素的重要性反映在不同組織學類型的免疫檢查點阻斷反應率的廣泛差異,以及具有相似分子和遺傳特征的腫瘤的高反應率(例如,微衛星不穩定性)。這些腫瘤的內在因素也會影響一些腫瘤細胞的外在因素(如宿主免疫系統和腫瘤相關基質)參與腫瘤治療臨床耐藥。
? ? ?? 在這篇綜述中,作者著重于腫瘤的內在因素對免疫檢查點阻斷的耐藥性。并回顧了腫瘤對免疫檢查點阻斷反應的免疫學基礎,重點介紹了關鍵的生物標志物,并討論了這些標志物是如何反映促進對免疫檢查點阻斷反應的腫瘤內在因素的。同時,作者研究腫瘤固有缺陷導致免疫檢查點阻斷的機制,并強調現有的和正在出現的克服腫瘤固有耐藥機制的方法。
一、腫瘤內在的抗性機制
? ? ?? 決定自然產生的抗腫瘤T細胞反應的誘導和維持的因素是復雜的。腫瘤細胞自身固有的特征,如腫瘤細胞突變譜,干擾素信號通路的功能,抗原遞呈分子的表達和免疫逃逸的致癌信號通路等,都會影響T細胞對腫瘤微環境的啟動、活化和募集,這個過程是在免疫檢查點阻斷的情況下,對免疫應答所必要的。同樣地,對免疫檢查點阻斷的抵抗力可能是由這些關鍵腫瘤特征的破壞所引起的,可以通過預防從頭進行的抗腫瘤免疫反應或通過抵消正在進行的抗腫瘤反應來實現。
二、腫瘤的抗原性不足
? ? ?? 多項研究表明,腫瘤新抗原有可能成為抗腫瘤免疫的有效靶標,而且突變負荷與跨惡性腫瘤對免疫檢查點阻滯的反應之間存在相關性。在對抗CTLA4免疫檢查點阻斷有反應的患者中,已顯示對特定腫瘤新抗原具有特異性的T細胞先前已存在于腫瘤微環境中,并在抗CTLA4療法的反應下擴增。在小鼠甲基膽堿誘發的肉瘤模型中,針對新抗原的T細胞可擴展并獲得抗腫瘤功能,以應對免疫檢查點阻斷。在沒有免疫檢查點阻斷的情況下,甚至可以在腫瘤微環境中檢測到強新抗原特異性T細胞。在患有轉移性膽管癌的患者中,腫瘤浸潤淋巴細胞帶有對腫瘤新抗原具有特異性的CD4 + T細胞群。過繼轉移富集的突變特異性T細胞導致有效的抗腫瘤反應。越來越多的證據,包括正在進行的基于新抗原的腫瘤疫苗研究的早期結果表明,新抗原是關鍵的癌癥免疫原。因錯配修復缺陷而導致微衛星不穩定的患者對免疫檢查點阻斷的反應率較高的觀察結果進一步支持了新抗原在抗腫瘤免疫反應中的作用。相反,抗原性差的腫瘤不太可能對免疫檢查點阻斷具有內在敏感性。
三、腫瘤固有白介素-γ信號通路
? ? ?? 針對腫瘤抗原的有效T細胞反應導致腫瘤微環境中干擾素γ(IFNγ)的表達,從而激活JAK和STAT信號轉導,從而誘導PD-L1表達。腫瘤細胞對IFNγ信號的反應中斷可阻止PD-L1表達的誘導,從而使PD1/PD-L1阻斷無效(圖1)。然而,早就知道破壞腫瘤細胞對IFNγ信號傳導的反應不僅是對免疫檢查點阻斷的抗性機制,而且更廣泛地是對抗腫瘤免疫的抗性機制。經過工程改造以表達顯性陰性IFNγ受體的小鼠腫瘤表現出更大的致瘤性,并且對通過全身性給予脂多糖引起的抗腫瘤免疫具有抵抗力。這些腫瘤也可以在對親代腫瘤具有事先免疫力的小鼠體內建立。當在缺乏IFNγ受體的小鼠中出現的自發性腫瘤被重新植入具有免疫能力和免疫缺陷的小鼠中時,它們以相似的動力學生長。但是,這些腫瘤中IFNγ受體的重組導致它們在具有免疫能力(但不是免疫缺陷)的小鼠中被排斥,這突顯了腫瘤固有的IFNγ信號通路在免疫排斥中的關鍵作用(圖2)。
? ? ?? 與IFNγ受體信號傳導在調節腫瘤的免疫原性中的作用相反,還提出了腫瘤細胞中的長期IFNγ受體信號傳導可以介導對免疫檢查點阻斷的抗性。這是基于抗病毒免疫的概念,其中長時間暴露于I型干擾素信號傳導會對病毒控制產生有害影響。一項研究表明,小鼠黑素瘤細胞在體外或體內長期暴露于IFNγ會導致PD-L1依賴性機制,其機制是通過上調替代性T細胞抑制性受體來實現對免疫檢查點阻斷的適應性抵抗,這是與IFNγ信號有關的表觀遺傳和轉錄組變化,尤其是STAT1。正在進行JAK抑制劑與抗PD1檢查點阻斷劑結合的臨床研究(NCT02646748和NCT03012230),但早期結果并不令人滿意。
? ? ?? 尚不清楚IFNγ信號傳導的下游功能中的哪一個對免疫檢查點阻斷的成功最關鍵。IFNγ信號傳導具有直接的抗增殖作用,導致抗原加工機制與表面MHC I類和II類分子的協同表達,并導致化學引誘劑如CXCL9和CXCL10的表達(圖2,3a)。源自人類黑色素瘤的細胞系在IFNγ信號傳導中具有固有的遺傳缺陷,不再對其抗增殖作用敏感,也不再上調MHC I類分子。
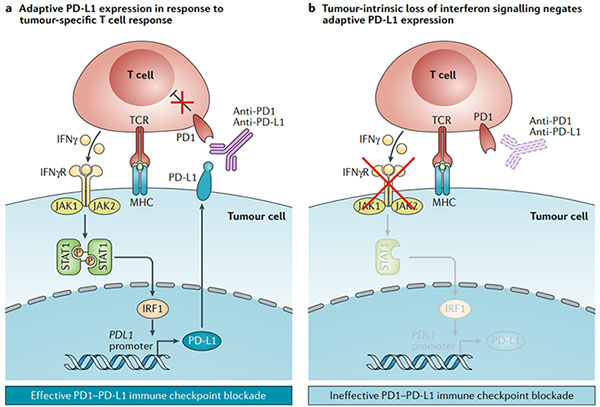
圖1.干擾素信號轉導自適應程序性細胞死亡1配體1表達
? ? ?? 預先存在的抗腫瘤免疫反應是有效的免疫檢查點阻斷的關鍵。在MHC I類或II類背景下識別腫瘤新抗原的腫瘤反應性T細胞釋放干擾素γ(IFNγ),從而導致Janus激酶(JAK)信號轉導子和轉錄激活子(STAT)信號通路的激活。這激活了轉錄因子干擾素調節因子1(IRF1),然后激活了PDL1的轉錄。這導致腫瘤細胞表面上程序性細胞死亡1配體1(PD-L1)的適應性表達,從而負調節抗腫瘤T細胞反應。針對PD1或PD-L1的抗體破壞了這種負反饋回路,以恢復抗腫瘤免疫力。b.類似的情況是,在MHC的情況下,腫瘤特異性T細胞遇到抗原,導致IFNγ釋放。然而,由于IFNγ信號傳導途徑中的遺傳缺陷(例如影響JAK1或JAK2),因此腫瘤細胞不會傳遞IFNγ信號,并且不會發生適應性PD-L1表達。在缺乏適應性PD-L1表達的情況下,PD1 PD-L1免疫檢查點阻斷無效。IFNγR,IFNγ受體;TCR,T細胞受體。
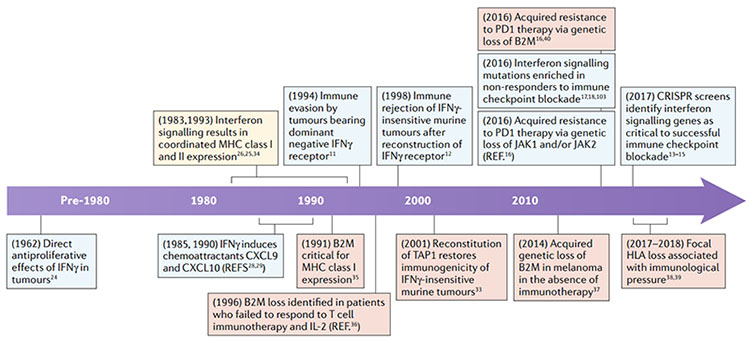
圖2.干擾素-γ(IFNγ)途徑和抗原呈遞在抗腫瘤免疫中的重要發現時間表
四、腫瘤固有的MHC缺失
? ? ? 腫瘤細胞可以通過下調表面MHC表達來逃避T細胞的殺傷。由于腫瘤抗原呈遞主要通過I類MHC途徑發生,因此與II類MHC抗原呈遞缺陷相比,該途徑的缺陷更為常見。然而,已經有人提出,黑色素瘤細胞上II類MHC的表達可能是抗PD1治療反應的生物標志物,并且可能受一組獨特的耐藥機制支配。
? ? ?? IFNγ信號傳導在抗腫瘤免疫中的重要作用可能與它誘導或增強MHC I類抗原呈遞的事實有關,這一過程需要幾個基因的協同表達,包括TAP1,TAP2,B2M和免疫蛋白酶體基因PSMB8,PSMB9 和PSMB10(圖3b)。對干擾素信號傳導缺乏敏感性的腫瘤細胞可能很少或沒有MHC I類抗原呈遞,從而允許免疫逃逸。在2001年的一項研究中,將TAP1穩定轉染到IFNγ缺陷的腫瘤細胞中導致它們在野生型但不是T細胞缺陷(Rag2- /)小鼠中被排斥(圖2)。確實,某些MHC I類缺陷的腫瘤細胞需要用IFNγ預處理以協調表達抗原加工機制和MHC I類肽復合物。
? ? ?? 即使在存在IFNγ信號的情況下,抗原加工設備中的缺陷也會破壞MHC I類表面表達(圖2,3b)。具有這種突變的腫瘤不僅對T細胞介導的免疫療法有抵抗力,而且這些突變實際上可能是免疫系統選擇性壓力的結果。例如,據報道接受免疫治療的黑色素瘤患者可能會失去β2-微球蛋白的功能性表達(B2M;從而喪失MHC I類表達)(圖2)。另一名轉移性黑色素瘤患者的縱向活檢標本顯示,在缺乏免疫療法的情況下,由于B2M丟失,導致獲得了MHC I類缺陷。最近的一種計算HLA拷貝數的計算方法允許研究人員推斷HLA基因座的克隆和亞克隆雜合性喪失的程度,雜合性事件的平行,亞克隆和局灶性HLA頻繁丟失,在轉移部位富集,表明存在免疫學壓力即使不進行免疫治療,在這些腫瘤中也存在。微衛星不穩定的結直腸癌患者具有很高的免疫原性,在免疫壓力和抗原加工機器的基因改變之間也有類似的關聯。
? ? ?? 毫不奇怪,已經報道了幾例獲得性抵抗免疫檢查點阻斷的耐藥性的案例,這些案例中編碼抗原加工機制的基因特別是B2M發生了突變。此外,B2M基因座雜合性的喪失與接受免疫檢查點阻斷治療的兩個獨立的黑色素瘤患者隊列的總體存活率降低相關。還已經確定了調節抗原呈遞的新基因。例如,體外功能獲得性激酶組篩選顯示,編碼HLA-A轉錄后負調節劑的MEX3B,可使黑素瘤細胞逃避腫瘤特異性T細胞(圖3b)。值得注意的是,抗PD1治療后MEX3B表達豐富了無反應的患者人群。
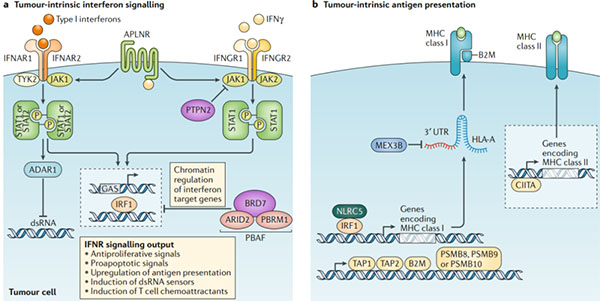
圖3.抵抗免疫檢查點阻斷:腫瘤內在逃逸機制
? ? ?? a.多個無偏倚的基于CRISPR的篩選發現了腫瘤內源性干擾素信號傳導對免疫檢查點阻斷和基于T細胞的免疫療法的關鍵作用。這些研究已經確定了干擾素-γ(IFNγ)和I型干擾素信號傳導途徑的組成部分。例如Janus激酶1(JAK1),JAK2,信號轉導和轉錄激活因子1(STAT1)以及IFNγ受體I(IFNGR1)和IFNGR2對免疫檢查點阻斷的成敗至關重要。這項研究還確定了鮮為人知的調節劑對免疫檢查點阻斷反應的作用,包括表面受體apelin受體(APLNR),該受體調節上游對IFNγ和I型干擾素信號傳導,酪氨酸蛋白磷酸酶非受體2型(PTPN2)的敏感性。它調節對IFNγ信號,BRD7和DNA結合亞基ARID2和PBRM1的上游敏感性,它們是染色質重塑復合物PBAF的一部分,并參與IFNγ靶基因的調控。以及雙鏈RNA(dsRNA)特異的腺苷脫氨酶(ADAR1),它對內源性dsRNA含量有負調節作用。I型干擾素和IFNγ信號在DNA中的IFNγ激活位點(GAS)上會聚,并激活轉錄調節因子,如干擾素調節因子1(IRF1),然后驅動干擾素信號傳導的關鍵輸出。
? ? ?? b.大量研究表明,抗原呈遞中與干擾素無關的缺陷也會導致免疫逃逸和對免疫檢查點阻斷的抵抗。這些缺陷可能發生在HLA基因座或MHC I類復雜成分β2-微球蛋白(B2M)中。其他缺陷可能會在抗原加工機制中發生,例如膜結合轉運蛋白TAP1和TAP2以及免疫蛋白酶亞基PSMB8,PSMB9或PSMB10,或者在MHC I類的轉錄調控中(例如細胞質蛋白NLRC5)。MHC I類表達也可能在轉錄后水平受到影響。RNA結合蛋白MEX3B可以結合HLA-A轉錄物,導致其降解并降低MHC I類分子的表達。發現對檢查站阻斷無反應的黑色素瘤患者MEX3B上調。盡管在腫瘤細胞上更高的MHC II類抗原呈遞與對免疫檢查點阻斷的改善反應相關,但在對免疫檢查點阻斷產生抗藥性的情況下,尚未發現MHC II類基因或MHC II類轉錄激活因子CIITA的遺傳缺陷。IFNAR1,干擾素-α和β受體亞基1;IFNR,干擾素受體;UTR,非編碼區。
五、由致癌信號通路調節
? ? ?? 致癌信號通路可能與癌癥發展各個階段的腫瘤免疫力有關,包括腫瘤起始,生長,侵襲和轉移。這些腫瘤內在途徑在塑造腫瘤免疫原性和免疫微環境中的作用最近在其他地方綜述了。在這里,我們著眼于三種途徑,這些證據支持在腫瘤內源性對免疫檢查點的抗性中發揮作用,從而阻斷了WNT-β-catenin途徑,細胞周期蛋白依賴性激酶4(CDK4)-CDK6途徑和有絲分裂原激活的蛋白激酶(MAPK)途徑, 以及PTEN丟失引起的途徑。
? ? ?? WNT-β-catenin信號傳導。WNT-β-catenin信號傳導是一種進化保守的信號傳導途徑,涉及多種細胞過程,包括腫瘤發生和胚胎發生。典型的WNT-β-catenin信號傳導是通過WNT家族蛋白與細胞表面受體的結合而啟動的,從而激活信號轉導,從而導致β-catenin的核轉位和轉錄激活。最近,它已成為一種致癌信號通路,阻礙了從頭抗腫瘤免疫反應的啟動。這種觀點是從一項觀察發現的,即具有活躍的WNT-β-catenin信號傳導的黑色素瘤樣本中約有三分之一缺乏明顯的T細胞浸潤。具有活躍WNT-β-catenin信號傳導的黑素瘤細胞系可產生免疫抑制細胞因子,例如IL-10。最近,研究表明,體內黑色素瘤細胞產生的WNT-β-catenin信號傳導可通過破壞表達堿性亮氨酸拉鏈轉錄因子ATF-like 3(BATF3)的樹突狀細胞的募集而阻止引發抗腫瘤反應。其他研究表明,源自黑素瘤細胞的可溶性WNT激動劑WNT5A可以激活樹突狀細胞中的β-catenin信號傳導,從而導致代謝向氧化磷酸化和脂肪酸氧化的轉變,其特征在于吲哚胺2,3-二加氧酶1(IDO1)和過氧化物酶體增殖物激活受體-γ(PPARγ)分別促進免疫抑制。具體而言,色氨酸向犬尿氨酸的轉化是由IDO1催化的,IDO1是WNT5A誘導的信號傳導下游的轉錄靶標。這種代謝轉變促進了調節性T細胞的發育,同時抑制了效應T細胞的活性。在Braf V600E / Pten-//小鼠黑素瘤模型中,抑制這種代謝變化可增強抗PD1免疫療法的功效。
? ? ?? 一系列針對幾種不同類型癌癥的研究表明,增強的WNT-β-catenin信號傳導與缺乏內在免疫細胞浸潤的腫瘤之間存在聯系,而內在免疫細胞浸潤對免疫檢查點阻滯反應的可能性較小(也稱為免疫學上的冷腫瘤)。這項研究包括一項整合了《癌癥基因組圖集》中來自結直腸癌的基因組,轉錄組學和免疫組化數據的研究,以及其他有關免疫性冷卵巢癌,頭頸癌,膀胱癌和腺樣囊性癌的研究。另一項研究鑒定了絲氨酸/ 蘇氨酸蛋白激酶PAK4(一種WNT信號介導物)可豐富黑色素瘤患者的免疫性冷腫瘤,對抗PD1免疫檢查點阻斷無反應。在多種小鼠模型中,PAK4的基因缺失或藥理抑制導致對抗PD1治療的耐藥性逆轉。
? ? ?? CDK4-CDK6與細胞周期有關。早期的證據表明細胞周期調控與致癌轉化之間存在聯系,這是通過觀察到細胞周期蛋白A基因座上病毒整合與病毒整合,腺病毒致癌基因E1A與細胞周期蛋白A的關聯以及D型細胞周期蛋白在甲狀旁腺腫瘤中的過度表達而觀察到的。CDK4和CDK6與腫瘤發生特別相關,因為它們與D型細胞周期蛋白一起促進細胞周期從G1期發展到S期。被發現的十年后,小分子palbociclib成為第一個獲得美國食品和藥物管理局批準的CDK4 / CDK6抑制劑。自2017年以來,至少有四項研究強調了CDK4 / CDK6抑制對抗腫瘤免疫的影響。例如,研究表明,CDK4 / CDK6抑制劑abemaciclib與抗PD-L1療法聯合使用具有更大的抗腫瘤作用。在小鼠乳腺癌模型中比單獨使用任何一種藥物。該觀察結果歸因于腫瘤細胞增加的雙鏈RNA(dsRNA)分子的產生和感覺,這可能是由于對藥物的反應中DNA甲基轉移酶水平降低所致。腫瘤細胞通過模式識別受體的表達識別諸如dsRNA之類的危險信號,從而導致促炎基因(包括編碼干擾素和抗原呈遞機制的基因)過表達。在另一項有關人類T細胞,患者來源的離體培養物以及自發和異種移植小鼠癌癥模型的組合研究中,palbociclib或trilaciclib與抗PD1阻斷劑的組合比單獨使用任何一種藥物更有效。在這里,CDK4 / CDK6抑制作用對抗腫瘤免疫力的影響主要歸因于它們對T細胞的直接影響,盡管降低了它們的增殖能力,但導致更大的IL-2產生和增加的腫瘤浸潤。考慮到CDK4 / CDK6在T細胞功能中的作用,CDK4 / CDK6抑制抗腫瘤免疫的作用是否受到對腫瘤細胞致癌信號的直接影響,可能取決于每個模型系統中CDK4 / CDK6的相關性。
? ? ?? 來自接受免疫檢查點阻斷治療的患者的黑色素瘤樣品的單細胞轉錄組研究確定了由CDK4 / CDK6驅動的耐藥性程序。利用來自癌癥基因組圖譜黑素瘤隊列的大量RNA測序數據,鑒定了與T細胞排斥相關的腫瘤細胞的基因表達特征,該基因表達特征與富含對免疫檢查點阻斷具有抗性的腫瘤的基因重疊。研究作者稱這種重疊基因設定了抗藥性計劃。表達抗性程序的細胞系的藥理學篩選發現它們對CDK4 / CDK6抑制劑敏感。此外,在先前用于研究CDK4 / CDK6抑制作用對乳腺癌細胞和小鼠模型的影響的數據集中,抵抗程序因對CDK4 / CDK6抑制作用而受到抑制。CDK4 / CDK6通過使腫瘤抑制性視網膜母細胞瘤相關蛋白1(RB1)磷酸化而起作用,與此相一致,CDK4 / CDK6抑制抑制了兩種RB充足的黑素瘤細胞系中的耐藥性程序,但在RB不足的黑色素瘤細胞系中卻沒有。
? ? ?? MAPK信號傳導。MAPK信號通路可通過增加免疫調節細胞因子IL-6和IL-10的表達在癌癥免疫逃逸中發揮作用。該信號通路對腫瘤免疫狀態的影響在黑色素瘤中尤為重要,其中約一半的腫瘤攜帶MAPK BRAF突變,BRAF-V600E激活突變,而免疫檢查點阻斷是一線治療。維拉非尼是BRAF突變的抑制劑,已顯示出可增加黑色素瘤細胞對T細胞的細胞毒性作用的敏感性,而不會影響T細胞的增殖能力。
? ? ?? vemurafenib還可以通過依賴于活化突變BRAF-V600E的方式通過IFNγ受體和腫瘤壞死因子受體的協同信號傳導誘導細胞周期停滯。為了支持這一發現,較早描述的與腫瘤特異性T細胞共培養的B16腫瘤細胞的CRISPR篩選也顯示,針對MAPK途徑負調控子的CRISPR指導物對抗性腫瘤細胞的富集。在另一項使用PD1阻斷治療的患者的黑色素瘤標本的RNA測序研究中,從沒有表現出反應的患者中采集的腫瘤樣品鑒定了基因簽名,該簽名與先前發表的與MAPK抑制劑耐藥相關的簽名重疊。但是,這些數據必須謹慎評估,因為其他兩個大的轉錄組數據集并不能證實這些數據,這些數據集來自接受免疫檢查點阻斷治療的黑色素瘤患者的腫瘤。抑制BRAF還可以破壞免疫抑制因子的腫瘤內在表達。例如,BRAFV600E腫瘤顯示出細胞因子IL-6,VEGF和IL-10的表達增加,它們具有免疫抑制功能,部分原因是它們對樹突狀細胞功能的影響(例如IL-12和腫瘤壞死因子的產生)。
? ? ?? 由于與野生型BRAF細胞中MAPK通路的反常激活有關的毒性問題,使用免疫檢查點阻斷和用vemurafenib抑制突變BRAF與維拉非尼的聯合療法的開發停滯了。相反,研究人員轉向使用MAPK /細胞外信號調節激酶激酶(MEK)抑制劑,該抑制劑可抑制BRAF-V600E和野生型BRAF細胞中的MAPK途徑,或MEK和BRAF抑制劑的組合。在結腸癌的臨床前模型(CT26模型)中,抗PD1治療與MEK抑制相結合可長期控制腫瘤。同樣,MEK和BRAF抑制的組合增強了過繼性T細胞療法和抗PD1阻斷的療效。最近報道了三項結合MAPK信號傳導抑制劑和免疫檢查點阻滯的臨床研究。其中兩項研究使用了dabrafenib(一種BRAF抑制劑),trametinib(一種MEK抑制劑)和抗PD1抗體pembrolizumab,兩者均觀察到高緩解率(63%和73%),以及較高的3級或更高的毒性作用(分別為58%和73%)。第三項研究包括接受cobimetinib(一種MEK抑制劑),vemurafenib和抗PD-L1抗體atezolizumab組合的患者,其中72%達到了客觀緩解(完全緩解率為21%)。Cobimetinib和vemurafenib治療的導入期導致循環增殖CD4 + T細胞水平相對增加。
? ? ?? 腫瘤抑制因子PTEN缺失。盡管已經在一個世紀中描述了腫瘤抑制因子在腫瘤發生中的典型作用,但直到20世紀末,才發現PTEN丟失是一種常見的致癌事件。一項研究針對基于T細胞的免疫療法的功效,嚴格檢查了PTEN缺失在人黑素瘤和黑素瘤同系小鼠模型中的作用。在缺乏PTEN的情況下,無論是在體內還是體外,腫瘤細胞對腫瘤特異性T細胞的細胞毒性作用都更具抵抗力。PTEN表達還與對抗PD1療法的反應以及患者體內離體擴增的腫瘤浸潤淋巴細胞的更成功產量相關。其他研究也觀察到了PTEN丟失的類似影響:來自癌癥基因組圖集軟組織肉瘤數據集的RNA測序數據顯示,在患有以下疾病的腫瘤中,與T細胞浸潤和溶細胞活性相關的基因(例如編碼CD8α和粒酶B的基因)的表達下降。PTEN的缺失。此外,在對抗PD1免疫檢查點阻滯有部分反應的患者中,發現無反應的病灶具有PTEN74的缺失,表明PTEN缺失可能在抗藥性中發揮作用。值得注意的是,PTEN可以通過響應DNA病毒,RNA病毒,多肌苷酸:聚胞苷酸和脂多糖的模式識別受體的激活來輔助干擾素調節因子3(IRF3)的核轉運,從而響應病毒刺激來促進I型干擾素信號傳導。這可能與使用靶向模式識別受體的新藥克服免疫檢查點阻滯的抗藥性有關。
? ? ?? 考慮到PI3K被PTEN負調節并且是癌癥中通常失調的激酶,已經提出抑制磷酸肌醇3-激酶(PI3K)作為提高抗腫瘤免疫力的治療方法。然而,不同的PI3K同工型在癌細胞(PI3Kα和PI3Kβ)和免疫細胞(PI3Kδ和PI3Kγ)中具有活性。盡管抑制富含腫瘤細胞的PI3K同工型可以減少腫瘤的生長,但是大多數支持PI3K抑制在提高抗腫瘤免疫力中起作用的證據都是基于對PI3Kγ或PI3Kδ的抑制。例如,在缺乏功能性PI3Kγ或PI3Kδ的宿主中生長的野生型腫瘤以T細胞依賴性方式減慢了腫瘤的生長。巨噬細胞中的PI3Kγ激活可以激活免疫抑制轉錄程序,該程序以與腫瘤細胞無關的方式阻止抗腫瘤T細胞功能。然而,腫瘤固有的PI3K抑制作用是否會影響抗腫瘤免疫性尚不清楚。正在進行臨床研究(例如NCT02646748),以評估PI3K抑制劑和免疫檢查點阻斷對實體瘤患者的綜合影響。
? ? ?? 腫瘤去分化和干細胞化。腫瘤發生或腫瘤干細胞對傳統的細胞毒性療法有抵抗力。已有證據表明,腫瘤的去分化或干性也可能在抵抗基于免疫的療法中起作用。對抗PD1免疫檢查點阻斷有抵抗力的黑色素瘤患者的腫瘤進行了轉錄組學分析,確定了豐富的莖樣間充質基因簽名。在一名針對過繼轉移的靶向黑素細胞分化抗原1(MART1)的T細胞有反應的患者中,復發的腫瘤失去了MART1的表達,這是一種脫分化和免疫治療耐藥性的現象,在體外表現出來。其他研究表明,腫瘤啟動干細胞可能表達負調控分子,例如CD80,PD-L1(參考文獻81)和NKGD2。最后,WNT信號傳導(先前被描述為介導免疫療法抗性的致癌途徑)在腫瘤干和去分化中也具有眾所周知的作用。
六、腫瘤固有抵抗力的生物標志物
1. 新生腫瘤反應性T細胞
? ? ?? 與基于活化的腫瘤特異性T細胞的過繼轉移的方法不同,免疫檢查點阻斷可利用自然發生的抗腫瘤T細胞反應。幾項觀察表明,免疫檢查點阻斷的功效取決于預先存在的免疫反應。首先,黑色素瘤是一種以其固有的免疫原性而聞名的腫瘤,對免疫檢查點阻斷單一療法的反應率很高。黑色素瘤長期以來一直是研究免疫療法的模型惡性腫瘤,轉移性黑色素瘤患者體內離體擴增的腫瘤浸潤淋巴細胞的過繼性細胞轉移的臨床成功為黑色素瘤患者存在天然存在的腫瘤特異性T細胞提供了證據。
? ? ?? 先前存在的抗腫瘤免疫反應的最簡單指標可能是腫瘤微環境中存在T細胞。在接受抗PD1治療的黑色素瘤患者中,預處理活檢標本中T細胞的存在與對治療的反應相關,和侵入邊緣的CD8 + T細胞密度在一個小型驗證隊列中可預測反應。對PD1阻斷療法有反應的患者的基線標本在浸潤邊緣也具有較高水平的磷酸化STAT1表達。這表明對治療的反應不僅需要T細胞的存在,而且還需要產生IFNγ的活化T細胞的存在,這會引發一個信號級聯反應,從而導致相鄰腫瘤和基質細胞中STAT1的磷酸化。這支持了腫瘤固有的IFNγ信號傳導在上述對PD1阻滯的應答中的作用。在患有錯配修復缺陷的大腸癌患者中抗PD1治療(pembrolizumab)的研究和尿路上皮癌患者抗PD-L1治療的研究中也報告了類似的發現。
? ? ?? 但是,這種觀察并不普遍。在先前接受過抗CTLA4治療或未接受抗CTLA4治療的患者隊列中,基線處存在的腫瘤浸潤淋巴細胞與抗PD1治療反應無關,在其他隊列中也有例外。這可能部分與用于分析的預處理活檢標本的腫瘤異質性和選擇偏倚有關。在某些情況下,患者可能攜帶腫瘤特異性T細胞,但局部免疫抑制因子限制了這些克隆的浸潤和擴增。在其他情況下,腫瘤特異性T細胞可能存在于外周,而不存在于腫瘤微環境中。這表明,對于從頭抗腫瘤免疫應答的存在,腫瘤浸潤淋巴細胞的存在不是特別敏感的替代物。
2. PD-L1作為干擾素信號的標記
? ? ?? 在某些而非全部情況下,已顯示腫瘤微環境中免疫檢查點分子(例如PD-L1)的表達可預測對免疫檢查點阻斷的反應。但是,PD-L1表達并不一定表明已存在抗腫瘤免疫反應。一些PD-L1陽性腫瘤患者對治療無反應,一些PD-L1陰性腫瘤患者也可以從免疫檢查點阻斷中獲益。
? ? ?? PD-L1的表達主要受干擾素信號傳導途徑調節,該信號傳導途徑包括激酶JAK1和JAK2以及轉錄因子STAT1,STAT2和STAT3,以及轉錄激活因子IRF1。IFNγ甚至可以刺激PD-L1在腫瘤來源的外泌體上的表達,這也可以介導CD8 + T細胞的抑制。在這種情況下,腫瘤浸潤性T細胞與表達PD-L1的腫瘤和/或免疫細胞共存,阻斷PD1 PD-L1軸可能是有效的(圖1a),并進一步支持了PD1的作用。腫瘤內源性IFNγ信號傳導對PD1阻滯的反應。
? ? ?? I型干擾素信號傳導和II型干擾素(IFNγ)信號傳導都收斂以激活類似的下游基因靶標,例如PDL1。I型干擾素主要由髓樣細胞響應模式識別受體的激活而產生,而II型干擾素主要由T細胞在識別同源抗原時產生。因此,在基于T細胞的抗腫瘤免疫中,II型干擾素起著更為重要的作用。干擾素信號傳導途徑中的突變(尤其是II型干擾素信號傳導)或限制腫瘤特異性PD-L1表達的表觀遺傳和轉錄后機制可使PD-1 PD-L1免疫檢查點阻斷多余。
? ? ?? PD-L1表達也可以通過各種其他機制進行調節。這些機制包括基因過表達(例如PD-L1,PD-L2和JAK2的基因座的擴增,稱為PDJ amplicon104),表觀遺傳沉默,轉錄調控(例如,通過MYC,PTEN和低氧誘導因子1α),轉錄后調節(通過microRNA),翻譯后修飾(糖基化,磷酸化和泛素化)以及胞質和內體重定位。這些過程也會影響對PD1阻斷療法的反應。
3. 從腫瘤轉錄組學得到的經驗
? ? ?? 盡管采用了新興的多重方法,但基于免疫組化的評估腫瘤免疫狀態的方法受到其分析范圍的限制。因此,RNA測序和靶向基因陣列已遠遠超過了它們。這些努力與RNA解卷積算法的進步(例如溶細胞活性評分,MCP計數器,CIBERSORT和TIMER)并行,可以評估大塊腫瘤標本的免疫細胞組成。溶細胞活性評分是RNA解卷積技術中最簡單的一種,它使用顆粒酶A和穿孔素表達的幾何平均值來總結腫瘤的效應T細胞組成。較高的基線溶細胞活性評分與抗CTLA4免疫檢查點阻斷的反應相關,病毒防御基因表達特征也是如此。
? ? ?? 但是,基于大塊腫瘤RNA的免疫標記具有缺點。腫瘤異質性是在研究內部和研究之間獲得可重復的,一致的結果的障礙。在接受抗CTLA4預處理的一組患者中(未接受過抗CTLA4的未接受過治療的患者),其細胞溶解活性評分增加,并且抗PD1反應患者的基線腫瘤中病毒防御標記豐富,但沒有特定的免疫人群在基線時通過CIBERSORT RNA解卷積鑒定出的蛋白與反應顯著相關。在另一組接受抗PD1治療的患者中,基線溶細胞活性評分或干擾素信號與反應無關。為了克服批量腫瘤轉錄組學的障礙,單細胞RNA測序工作正在進行中。對來自32位接受免疫檢查點阻斷治療的患者的48份腫瘤活檢標本進行的分析顯示,在有反應的患者的基線標本中CD8 + T細胞浸潤(由免疫組織化學定義)沒有增加。然而,單細胞RNA測序顯示,與CD8 + T細胞相比,應答者的基線標本中的CD8 + T細胞富含與記憶細胞分化(例如,編碼轉錄因子的TCF7),激活和細胞存活有關的轉錄本。無應答者,富含與疲勞相關的基因。
4. 腫瘤新抗原作為T細胞靶向
? ? ?? 盡管有證據表明新抗原特異性T細胞反應是免疫檢查點阻斷功效的核心,但突變負荷有限,無法預測對免疫檢查點阻斷反應的反應。這可能部分是由于所討論的突變的克隆性。在患者的所有腫瘤細胞中共有的克隆突變對于產生有效的抗腫瘤反應可能更為關鍵。
? ? ?? 此外,為了使突變成為免疫學靶標,必須通過MHC抗原有效地將其呈遞給免疫系統。盡管新抗原預測工具已得到改進,但缺乏用于驗證這些預測工具的高通量檢測方法限制了其進展。例如,在接受抗CTLA4治療的一組患者中,預測的新抗原負荷沒有超過突變負荷作為反應的生物標志物。最后,鑒于腫瘤微環境中抗腫瘤免疫反應的其他障礙的存在,新抗原的存在可能是不足的生物標志物。實際上,在多個針對PD1治療的前瞻性研究中,既捕獲了腫瘤內在突變負擔又感染了腫瘤微環境的生物標志物與反應的相關性更強,而不是單獨使用任何一種生物標志物。
七、克服腫瘤固有抵抗力
? ? ?? 與單獨使用任一療法相比,將抗CTLA4和抗PD1阻斷劑聯合使用所見的優異的抗腫瘤反應表明,這兩個免疫學檢查要點具有非冗余的分子機制。轉錄組學和免疫組化數據表明,對雙重免疫檢查點阻滯的反應者是那些已經存在的生產性抗腫瘤反應,被免疫檢查點所抑制的反應超出了PD1 PD-L1和CTLA4的阻滯(圖4a)。幾種靶向替代性免疫檢查點的抑制劑處于臨床前和臨床開發階段,包括靶向LAG3,VISTA,TIM3,腺苷A2A受體,CD73,BTLA,B7-H3,B7-H4和殺傷細胞免疫球蛋白樣受體的抑制劑。
? ? ?? 但是,這使很大一部分患者具有免疫學上較冷的腫瘤,不太可能對單一或聯合免疫檢查點阻斷產生反應。對于這些患者,目的是通過增強抗原呈遞和引發針對現有抗原的免疫反應來啟動抗腫瘤免疫反應。腫瘤及其引流性淋巴結據稱是腫瘤抗原呈遞的主要部位,因此調節腫瘤內和淋巴結抗原呈遞的腫瘤導向方法引起了人們的興趣(圖4b)。這些方法基于:(1)在腫瘤微環境中誘導壓倒免疫抑制基礎機制的促炎狀態;(2)誘導免疫原性細胞死亡;(3)募集專業抗原呈遞細胞(APC)來有效引發針對腫瘤抗原的啟動。腫瘤內免疫刺激劑的一個早期實例是卡介苗(Calillus CalmetteGuérin),它是淺表性膀胱癌的標準療法。
? ? ?? 化學療法和放射療法都可以通過多種提議的機制誘導免疫原性細胞死亡,這些機制在其他地方有詳細介紹。在小鼠模型中,化學療法和放射療法的免疫效果均依賴于T細胞,并且兩者均可 增強免疫檢查站阻斷的影響。然而,化學療法和放射療法具有充分證明的免疫抑制功能,可誘導對免疫療法產生耐藥性的腫瘤外源性機制。因此,這些標準療法不太可能作為克服對免疫檢查點阻斷的內在抗性的主要方法出現,但是它們在控制疾病負擔和引發免疫原性細胞死亡中的作用可能與新興的聯合免疫療法結合起來有用。
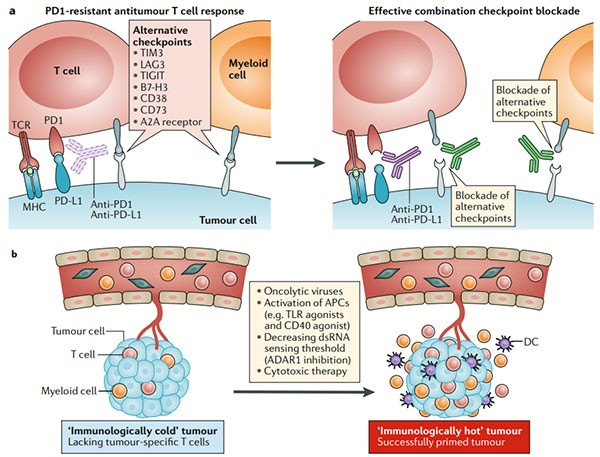
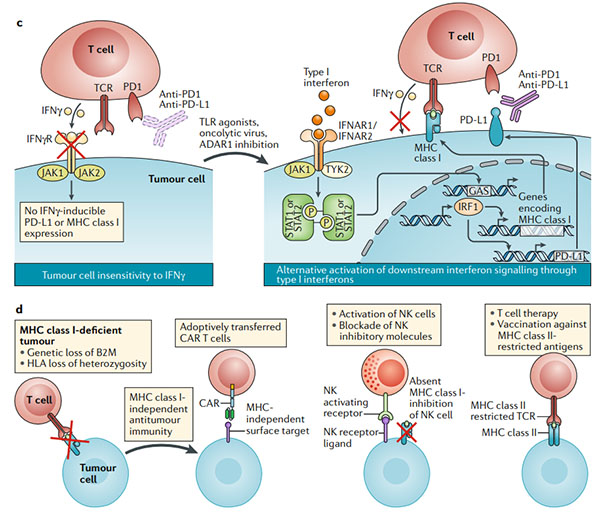
圖4.克服腫瘤對免疫檢查點阻斷的內在抗性
? ? ?? a.程序性細胞死亡1(PD1)PD1配體1(PD-L1)軸可能不是抗腫瘤T細胞反應的唯一負調節劑。在腫瘤細胞或腫瘤微環境中的髓樣細胞上表達的其他免疫檢查點分子會阻止有效的抗腫瘤免疫力;組合的免疫檢查點阻斷可能會破壞這種耐藥機制(右圖)。
? ? ?? b.免疫學上較冷的腫瘤類型缺乏預先存在的抗腫瘤T細胞反應,使免疫檢查點阻斷無效。通過引起免疫原性細胞死亡(溶瘤病毒或細胞毒性療法),引發抗原呈遞細胞(APC;使用Toll樣受體(TLR)激動劑和CD40激動劑)或增加腫瘤細胞對雙鏈RNA敏感性來引發針對腫瘤的免疫系統的方法(dsRNA;例如對dsRNA特異性腺苷脫氨酶(ADAR1)的抑制作用)可以將免疫寒冷狀態重新編程為檢查點阻斷反應狀態。
? ? ?? c.沒有干擾素-γ(IFNγ)信號傳導的腫瘤缺乏響應IFNγ自適應表達PD-L1的能力。在一些MHC I類抗原呈遞主要依賴于IFNγ信號傳導的腫瘤中,IFNγ信號傳導的喪失等同于抗原呈遞的喪失。通過TLR激動劑,溶瘤病毒或其他方式激活其他干擾素途徑(I型干擾素)也可能導致信號轉導子和轉錄激活因子1(STAT1)和STAT2信號激活,從而激活PD-L1和MHC的轉錄通過干擾素調節因子1(IRF1)的誘導達到I類。
? ? ?? d.其他腫瘤通過基因改變(例如,β2-微球蛋白(B2M)的喪失和HLA雜合性的喪失)喪失了I類MHC表達后,對免疫檢查點阻斷具有抵抗力。在這種情況下,三種方法可以成功:(1)嵌合抗原受體(CAR)T細胞獨立于MHC I類表達識別其靶標;(2)過繼轉移自然殺傷(NK)細胞或用細胞因子(例如IL-2或IL-15)刺激NK細胞,因為這些靶細胞缺乏MHC I類表達;(3)接種疫苗或過繼性T細胞療法產生針對特定MHC II類限制性抗原的反應。B2M,β2-微球蛋白;DC,樹突狀細胞;GAS,IFNγ激活位點;IFNAR1,干擾素-α和β受體亞基1;IFNγR,IFNγ受體;JAK1,Janus激酶1;TCR,T細胞受體。
? ? ?? 化學療法和放射療法都可以通過多種提議的機制誘導免疫原性細胞死亡,這些機制在其他地方有詳細介紹。在小鼠模型中,化學療法和放射療法的免疫效果均依賴于T細胞,并且兩者均可 增強免疫檢查站阻斷的影響。然而,化學療法和放射療法具有充分證明的免疫抑制功能,可誘導對免疫療法產生耐藥性的腫瘤外源性機制。因此,這些標準療法不太可能作為克服對免疫檢查點阻斷的內在抗性的主要方法出現,但是它們在控制疾病負擔和引發免疫原性細胞死亡中的作用可能與新興的聯合免疫療法結合起來有用。
? ? ?? 溶瘤病毒具有感染腫瘤細胞并誘導細胞死亡的獨特能力。為了治療目的,它們也經常被基因改造以增強抗腫瘤免疫反應。基于單純性1型皰疹病毒的Talimogene laherparepvec(或T-VEC,以Imlygic形式銷售)是第一種獲得美國食品藥品監督管理局批準的溶瘤病毒。它在轉移性黑色素瘤患者體內腫瘤內傳遞,在腫瘤細胞內優先復制并表達細胞因子GM-CSF以促進附近APC的成熟和激活。T-VEC被設計為不干擾被感染細胞中的抗原呈遞,這與其衍生的病毒載體不同。結合抗PD1治療,在轉移性黑色素瘤患者的Ib期研究中,T-VEC的客觀緩解率達到62%,比單獨抗PD1治療的預期緩解率更高。最值得注意的是,13例CD8 + T細胞浸潤低的患者中有9例具有客觀反應,而5例基線IFNγ產生低的患者中有3例具有完全反應,支持了T-VEC在沒有預先存在的抗腫瘤免疫反應的患者中的作用。提出的機制如下:雖然病毒介導的免疫原性細胞死亡導致肽抗原的可用性,但病毒抗原的先天傳感器促進IFNγ信號傳導。這與T-VEC強制表達GM CSF一起,導致在腫瘤微環境中募集和激活APC。然后,APC在腫瘤微環境或引流淋巴結中引發或激活腫瘤特異性T細胞,從而逆轉了由腫瘤建立的先前存在的免疫排斥。
? ? ?? 增強腫瘤免疫原性的非基于病毒的腫瘤定向方法包括模式識別受體的激活劑,例如SD-101,這是一種具有CpG基序的合成寡核苷酸,可激活腫瘤微環境內腫瘤和非腫瘤細胞上的TLR9信號傳導。在Ib期研究中,先前未接受過抗PD-1治療的78%黑色素瘤患者具有客觀反應。在多個小鼠模型中的臨床前研究支持使用TLR9激動劑CpG寡核苷酸誘導全身性抗腫瘤免疫。這些包括SD-101和OX40激動劑抗體的組合,在多種模型中有效,包括轉移性乳腺癌的自發小鼠模型。由于I型干擾素信號傳導的遺傳或表觀遺傳缺陷,固有免疫傳感器的激動劑(可能是I型干擾素信號傳導的強效誘導劑)也可以為腫瘤中的抗原呈遞提供刺激,而這些抗原可抵抗免疫檢查點阻斷。
? ? ?? 腫瘤細胞對免疫檢查點阻斷的敏感性可以通過其對內源性先天免疫信號(例如內源性dsRNA)的內在敏感性進行微調。將腫瘤細胞的設定點更改為內源性dsRNA(即降低dsRNA閾值)可能是克服腫瘤對免疫檢查點阻斷的內在抗性的途徑。在針對小鼠黑素瘤細胞中2300多個基因的體內CRISPR篩選中,對抗PD1和GVAX13的反應更好的腫瘤中,ADAR1的丟失增加了,ADAR1編碼一種將腺苷轉化為肌苷的RNA編輯酶。B16小鼠黑素瘤中ADAR1的缺失逆轉了腫瘤微環境的免疫寒冷狀態,并增加了腫瘤細胞對I型或II型干擾素直接抗腫瘤作用的敏感性142。在ADAR1缺陷型腫瘤中對抗PD1治療的改善反應取決于兩個dsRNA傳感器MDA5和PKR中至少一個的存在。這些數據支持了腫瘤內在RNA感應在免疫檢查點阻斷功效中的作用。然而,仍在研究腫瘤固有的細胞質DNA的作用。
? ? ?? 宿主先天免疫感應在免疫檢查點反應中起著眾所周知的作用,但在適當條件下可能是必需的。胰腺導管腺癌的T細胞浸潤最少,對免疫檢查點阻滯的反應較差。在胰腺癌小鼠模型中,化學療法,共刺激蛋白CD40的激動劑和抗PD1療法的組合可產生T細胞依賴性抗腫瘤功效。CD40在包括樹突狀細胞在內的免疫細胞中廣泛表達,已知其與CD40配體的結合可許可抗原呈遞。結合化學療法和抗PD1治療,CD40激活和化學療法誘導的免疫原性細胞死亡以BATF3 +樹突狀細胞依賴性方式驅動T細胞活化,但獨立于宿主先天性免疫信號傳導途徑,包括通過MYD88,TLR4, TRIF,TLR3,STING,P2X74,caspase 1和caspase11。CD40激動劑聯合化學療法和抗PD1療法的I / II期研究正在進行中(NCT03214250)。
? ? ?? 對于攜帶遺傳缺陷會損害MHC I類或II類抗原呈遞的腫瘤的患者,正在研究至少兩種基于免疫的方法(圖4d)。基于嵌合抗原受體(CAR)的過繼T細胞療法是一種針對血液系統惡性腫瘤的有效免疫療法,由于它直接靶向腫瘤細胞表達的特定表面分子,因此無需通過MHC提呈抗原。然而,由于缺乏腫瘤特異性表面抗原和免疫抑制性微環境,因此對于CAR T細胞而言,成功治療實體瘤一直是遙不可及的。新穎的工程方法可創建雙重靶標活化的CAR T細胞,具有合成AND門邏輯開關以提高安全性和靶標特異性的CAR T細胞以及對腫瘤微環境中的免疫抑制信號不敏感或協同選擇的CAR T細胞(例如轉化生長因子-β信號轉導)有望為該方法提供前景。針對MHC缺乏型腫瘤的另一種方法是使用自然殺傷(NK)細胞進行細胞療法,以消除缺乏MHC I類分子的細胞。缺乏對ADAR的B2M缺陷型B16腫瘤,對GVAX和抗PD1療法的聯合治療敏感,被發現會增加NK細胞的浸潤。NK細胞療法的研究已經進行了數年的研究,最近,NKG2A的阻滯在NK和T細胞上均表達,這是一種基于酪氨酸的抑制性基序,已被證實在頭頸部鱗狀細胞癌患者中具有活性。
? ? ?? 最后,為了解決由致癌信號驅動的免疫檢查點阻斷抗性的機制(圖5a),研究人員將致癌信號通路的現有抑制劑重新定位為增強抗腫瘤免疫力的一種方法(特別是與免疫檢查點阻斷結合使用)。這些包括WNT信號抑制劑,CDK4和CDK6抑制劑以及MAPK和PI3K抑制劑(如圖5b所示)。
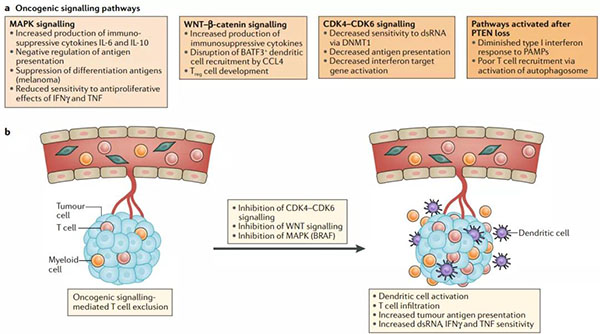
圖5.影響抗腫瘤免疫力和免疫檢查點阻滯性的致癌信號通路
? ? ?? a.致癌信號通路提供了獨特的免疫逃逸的腫瘤內在機制。在這里,我們重點介紹了與抗腫瘤免疫有關的四個關鍵致癌信號通路:有絲分裂原活化蛋白激酶(MAPK)信號通路,WNTβ-連環蛋白通路,細胞周期蛋白依賴性激酶4(CDK4)CDK6細胞周期信號通路和激活的 磷酸肌醇磷酸酶PTEN丟失的結果。
? ? ?? b.CDK4 CDK6信號傳導(例如使用palbociclib或abemaciclib),MAKP信號傳導(BRAF抑制劑)或WNT信號的治療性破壞可逆轉腫瘤固有T細胞的排除狀態,并恢復對免疫檢查點阻斷的敏感性。BATF3,基本的亮氨酸拉鏈轉錄因子ATF樣3;CCL4,CC趨化因子配體4;DNMT1,DNA(cytosine-5)-methyltransferase 1; dsRNA,雙鏈RNA;IFNγ,干擾素-γ;PAMPs,病原體相關分子模式;TNF,腫瘤壞死因子;Treg細胞,調節性T細胞。
Conclusion
? ? ?? 識別對免疫檢查點阻滯的抗性機制的過程已經重新發現了調節抗腫瘤免疫的主要機制。腫瘤對免疫檢查點阻斷的敏感性由腫瘤生物學決定:具有共同的組織學、分子和遺傳特征的腫瘤患者對免疫檢查點阻斷的反應率相似。腫瘤內源性因子通過其對腫瘤與宿主免疫系統之間相互作用的影響,可以間接在抗腫瘤外源性機制中發揮作用。但是,本綜述的重點放在直接受腫瘤內在因素影響的耐藥機制上。由于增加了突變負荷和抗原性,激活了從頭抗腫瘤免疫反應的腫瘤最有可能受益于免疫檢查點的阻斷。但是,即使具有足夠的抗原性,IFNγ信號傳導和抗原呈遞中的腫瘤固有遺傳缺陷也會破壞對免疫檢查點阻斷的敏感性。致癌信號通路通過指示募集對于啟動和影響抗腫瘤免疫應答至關重要的細胞,影響IFNγ和抗原呈遞通路,或者通過在腫瘤微環境中誘導免疫抑制因子,也是免疫檢查點阻斷抗性的介質。繞過IFNγ信號傳導和抗原呈遞缺陷或抑制免疫抑制致癌信號通路的靶向方法有望擴大免疫檢查點阻斷的影響。
? ? ?? 如涉及知識產權請與我司聯系
文獻來源:
Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25–39. doi:10.1038/s41577-019-0218-4
|


